Рентгеновская, электронная и ЯМР-кристаллография для определения структуры малых органических молекул
НОВОСТИ JEOL, том 53, № 6
Юсуке Нишияма1, 2
1 ДЖЕЛ РЕЗОНАНС Inc. 2 Центр сотрудничества RIKEN CLST-JEOL
Здесь мы представляем недавнюю разработку совместной лаборатории RIKEN CLST-JEOL по изучению молекулярных структур низкомолекулярных фармацевтических соединений в естественном изобилии (без какой-либо изотопной маркировки); Недавний прогресс в технологии быстрого вращения под магическим углом (MAS) в твердотельном ядерном магнитном резонансе (ssNMR) и в камере сверхвысокой чувствительности в просвечивающей электронной микроскопии (TEM) прокладывает новый путь для решения проблем в фармацевтической промышленности и науке. 1) кристаллические полиморфы и 2) соль/сокристалл являются двумя основными проблемами с точки зрения контроля качества, стабильности и интеллектуальной собственности. Для определения кристаллической формы проводят порошковую рентгенографию и 13C кросс-поляризационный MAS ssNMR широко используется, однако первый иногда не подходит для анализа смесей, а второй не позволяет различить кристаллические формы с похожими молекулярными конформациями. Для решения этих вопросов мы используем дифракцию электронов (ЭД) и 1H быстрый MAS ЯМР. Кристаллическую форму можно определить от монокристаллов размером от нано до микрометра с помощью ЭД, поскольку взаимодействия электронов на 4-5 порядков сильнее, чем взаимодействия рентгеновских лучей. 1H ЯМР также дает подходящую информацию о молекулярной упаковке, поскольку 1Н расположен на поверхности кристаллов. Проблема соли/сокристалла, где водород играет ключевую роль, является серьезной проблемой, поскольку рентгеновская дифракция монокристалла (SCXRD) не может точно определить положение атома водорода. Здесь мы определяем межъядерные расстояния между 1Рука 15N с использованием ssNMR в быстрых условиях MAS, в то время как глобальная структура получена с помощью SCXRD, отвечая на вопросы о соли / сокристалле.
Введение
В то время как лекарства на основе биотехнологий занимают лидирующие позиции на фармацевтическом рынке, традиционные низкомолекулярные препараты по-прежнему очень важны для ежедневного лечения, например, болезней взрослых. Эти низкомолекулярные активные фармацевтические ингредиенты (АФИ) обычно могут кристаллизоваться в нескольких различных формах, т.е. кристаллических полиморфах, в зависимости от условий кристаллизации. Поскольку на растворимость и стабильность в значительной степени влияет кристаллическая форма, очень важно контролировать и контролировать кристаллическую форму с точки зрения контроля качества от разработки до стадии производства [1, 2, 3]. Когда доступны кристаллы достаточно большого размера (~ 100 мкм), монокристаллическая рентгеновская дифракция (SCXRD) дает отчетливый ответ на кристаллические формы с атомарным разрешением. Однако большинство низкомолекулярных препаратов выпускается в микрокристаллических формах с различной рецептурой, включая таблетки, пилюли, порошки. Поскольку эти составы включают эксципиент, крайне важно определить кристаллическую форму по микрокристаллам в смеси. Порошковая рентгеновская дифракция (PXRD) и 13Ядерный магнитный резонанс с вращением под магическим углом с кросс-поляризацией (CPMAS ЯМР) - это два основных метода идентификации кристаллической формы. Поскольку экспериментальные картины/спектры дают отпечатки пальцев каждой кристаллической формы, сравнение картин/спектров между лекарственным средством и стандартной формой дает в большинстве случаев четкий ответ на кристаллическую форму в нем. Однако оба метода все еще имеют практические проблемы. PXRD иногда не может идентифицировать кристаллическую форму, так как многие дифракционные картины от API и наполнителя накладываются друг на друга. С другой стороны, 13C СРМАС, который чувствителен к молекулярной конформации, является подходящим методом для анализа смесей, поскольку большинство сигналов от эксципиента появляются в положениях, отличных от тех, что от АФИ, что позволяет избежать перекрытия сигналов. Однако, 13C CPMAS довольно нечувствителен к молекулярной упаковке, так как атомы углерода погружены внутрь молекул и расположены далеко от молекулярной поверхности. Таким образом 13C CPMAS не может идентифицировать кристаллические формы со сходными молекулярными конформациями. Более того, чувствительность к кристаллической форме меньше, чем у PXRD.
Другой важной проблемой низкомолекулярных АФИ является способ идентификации соли/сокристалла/континуума в многокомпонентных системах. Иногда требуется улучшить растворимость и/или стабильность кристаллов АФИ. Многокомпонентные системы, состоящие из АФИ и инертного соформера, являются одним из широко используемых решений этой проблемы. Многие примеры можно найти в многокомпонентных системах путем смешивания (обычно основного) АФИ и (кислотного) сообразователя. Когда разница pKa (ΔpKa) больше 3, образуется соль, в которой обнаруживаются межмолекулярные ионные взаимодействия. В соли протон из соформера полностью движется в сторону АФИ. В последнее время вводится еще один класс многокомпонентных систем — сокристаллы, когда ΔpKa меньше 3. В сокристалле протон в соформере все еще остается и образуется межмолекулярная водородная связь. Кроме того, встречается и система между солью и сокристаллом, т.е. континуум, в котором протон находится между АФИ и соформером. Крайне важно идентифицировать соль/сокристалл/континуум с интеллектуальной точки зрения, особенно когда ΔpKa меньше 3. Однако отсутствие возможности определения положения протона в методах, основанных на XRD, создает серьезную проблему для идентификации соли/сокристалла/континуума. континуума, так как различие между ними происходит только по водородным позициям.
Основными проблемами, которые еще не решены, являются 1) анализ смеси, 2) идентификация кристаллической формы со сходной молекулярной конформацией и 3) отсутствие возможности определить 1позиции Н. Первые два вопроса связаны с кристаллическими полиморфами, а последний — с солью/сокристаллом/континуумом. Здесь мы объединяем дифракцию электронов (ЭД), 1H ssNMR и SCXRD для решения этих проблем. ЭД является одним из методов наблюдения в оборудовании для просвечивающей электронной микроскопии (ПЭМ) и дает дифракционные картины. Поскольку взаимодействие электронов в 104–105 раз сильнее, чем взаимодействие рентгеновских лучей, картину ЭД можно наблюдать в монокристаллах от нано- до микроразмеров, что позволяет проводить анализ смесей. Хотя применение ЭД в значительной степени ограничивается неорганическими материалами, устойчивыми к электронному облучению, недавний прогресс в высокочувствительной камере позволяет работать в режиме низкой дозы вместе с криодержателем образца, открывая путь к ЭД-наблюдению органических кристаллов, включая низкомолекулярные. API веса. Вопрос 2) поднят потому, что 13C CPMAS нечувствителен к молекулярной конформации, как упоминалось выше. К счастью, внедрение очень быстрой технологии MAS > 70 кГц позволяет использовать высокое разрешение. 1H наблюдается даже в твердых телах [4, 5, 6], где 1Hs тесно связаны с 1H-1H-диполярные взаимодействия при умеренной скорости MAS. С 1H находится на поверхности, 1H-изотропные химические сдвиги чувствительны не только к конформации, но и к молекулярной упаковке. Кроме того, 1H-1Межмолекулярная корреляция H дает четкие закономерности для каждой молекулярной упаковки. Быстрая технология MAS реализует не только высокое разрешение 1H ЯМР, но и несколько сложных экспериментов, в том числе 14N ЯМР и 1H-15N измерений расстояния. Первый обнаруживает протонированное состояние в небольших органических молекулах. Последнее дает четкий ответ на проблему соли/сокристалла/континуума, где ключевую роль играют положения водорода. Кроме того, мы оцениваем пропускную способность каждого метода.
Кристаллические полиморфы[7]
Идентификация кристаллических форм в фармацевтических применениях имеет решающее значение и обычно выполняется с помощью PXRD и 13C CPMAS ssЯМР. Однако первый не подходит для анализа смесей, а второй иногда не работает, как обсуждалось выше. В качестве примера, 13C CPMAS спектры трех различных (псевдо) полиморфов L-гистидина показаны на рис.2O (рис. 1а) из-за различной молекулярной конформации. С другой стороны, L-гистидин в орторомбической (рис. 1б) и моноклинной (рис. 1в) формах дает практически одинаковые 13C CPMAS-спектры, отражающие близкую конформацию друг к другу. Как показано здесь, 13C CPMAS является чувствительной мерой для идентификации кристаллической формы с различной конформацией, однако не позволяет различить полиморфы со сходной молекулярной конформацией. Здесь мы предлагаем комбинированный подход ЭД и 1H ssNMR при очень быстром MAS, чтобы ответить на эти вопросы.
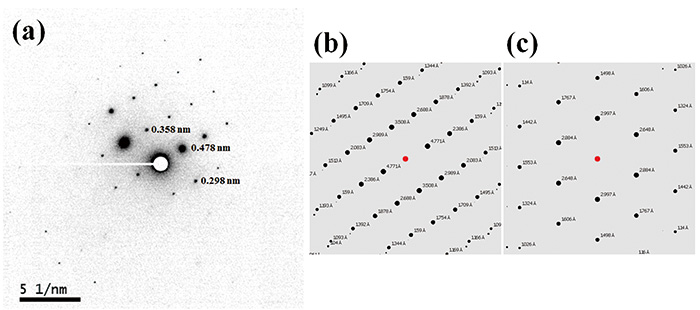
На рис. 2 представлены ПЭМ-изображение и картина ЭД L-гистидина·HCl·H.2О измерено при комнатной температуре. В то время как размер кристалла составляет около 1 мкм, что обычно встречается в фармацевтических продуктах, применяется нанолуч 100 нм. Таким образом, кристаллы размером 100 нм в принципе достаточно велики для получения монокристаллических картин ЭД. На самом деле, мы измеряли картины ЭД от кристаллов размером 100 нм или даже меньше. Критическая доза L-гистидина·HCl·H2O при комнатной температуре составляет всего 10-20 e-nm-2, устанавливаем мощность дозы 10 e-nm-2s-1 при времени экспозиции 1 с, в результате общая доза 10 е-nm-2. Даже в условиях такой очень низкой дозы современная CMOS-камера (OneView, Gatan, Inc., США) обеспечивает достаточную чувствительность. В то время как критические дозы 10-20 е-nm-2 обычно обнаруживаются в органических молекулах, при необходимости держатель для криопроб дополнительно улучшает критическую дозу. Поскольку картину ЭД можно легко рассчитать по кристаллической структуре, кристаллическую форму можно определить путем сравнения экспериментальной и рассчитанной картин ЭД. ЭД L-гистидина в орторомбической и моноклинной формах показаны на рис. 3 и 4 соответственно. Пока 13C CPMAS не может различить эти две формы (рис. 1), ED-дифракция дает разные картины для каждой кристаллической формы. Это связано с тем, что параметры решетки в этих двух формах совершенно разные. Кроме того, ED обеспечивает высокую производительность измерений в течение одной минуты по сравнению с PXRD (десятки минут) и 13C CPMAS (часы). Измерения ЭД проводили с помощью JEM-ARM200F (JEOL Ltd., Япония). Большинство приборов TEM способны выполнять это измерение, если установлена камера с высокой чувствительностью.
Молекулярную упаковку можно исследовать с помощью 1H ЯМР при очень быстром MAS. 1D 1Спектры ЯМР H полиморфов L-гистидина (псевдо) при 70 кГц MAS показаны на фиг. 5, что дает разные картины. Примечательно, что L-гистидин в орторомбической форме (b) дает структуру, отличную от таковой в моноклинной форме (c). Эти различия происходят из-за разных молекулярных упаковок. Это подчеркивает преимущество 1H ЯМР над 13C CPMAS, который не может идентифицировать эти две кристаллические формы. Благодаря высокому содержанию (> 99%) и ларморовской частоте (600 МГц при 14.1 Тл) для наблюдения этих одномерных спектров требуется менее одной минуты. Разницу между двумя кристаллическими формами можно усилить, наблюдая 1H/1H эксперименты по гомоядерной корреляции, которые должны быть чувствительны к межмолекулярным 1H/1H связность, таким образом кристаллические упаковки. Способность к гомоядерной корреляции является одной из уникальных особенностей 1H ЯМР с помощью его высокой чувствительности и изобилия. Рисунок 6 дает 2D 1H двухквантовый (DQ) / одноквантовый (SQ) гомоядерный корреляционный спектр L-гистидина в орторомбической и моноклинной формах. В то время как одномерные спектры отображают только локальную информацию, двухмерные гомоядерные корреляционные спектры также отражают информацию о молекулярных упаковках, что ясно показано на наложенных спектрах. Поскольку когерентность DQ создается посредством 1H-1При дипольных взаимодействиях интенсивности перекрестных пиков отражают пространственную близость. Например, межъядерные расстояния между H1 и H4 составляют 4.1 Å для орторомбической формы, значительно короче и 2.86 Å для моноклинной формы. Хотя разница в расстоянии составляет всего 1.4 раза, это приводит к усилению дипольного взаимодействия в 2.9 раза в моноклинной форме. Таким образом, корреляции H1-H4 наблюдаются только в моноклинной форме. Это 2D-эксперимент, однако время измерения обычно составляет менее одного часа, что дает более высокую пропускную способность, чем 13С КПМАС.
1H/14Корреляция N ЯМР также дает полезную информацию. В L-гистидине существует несколько возможных протонированных состояний. Любой или оба из двух атомов азота при τ и δ в имидазольном кольце могут быть протонированы до фрагмента NH. Кроме того, L-гистидин может быть цвиттерионом. Трудно различить эти протонированные состояния с помощью XRD или ED, поскольку оба метода не чувствительны к положениям водорода. Однако, 1H/14N корреляционных спектров L-гистидина (рис. 7) дают четкий ответ. В кислой среде в L-гистидин·HCl·H2O, появляется корреляция трех NH. Это ясно показывает, что атомы азота τ и δ протонированы. Еще одна корреляция NH появляется в низкочастотной позиции в 14Размер N при -260 ppm предполагает, что эта аминокислота представляет собой цвиттерион с NH3+ часть. Это связано с малой квадрупольной связью в NH3+ из-за локальной симметрии. С другой стороны, два полиморфа L-гистидина дают только два пика NH для каждого. Ясно, что оба являются цвиттер-ионами, и только один из азота τ и δ протонирован. Следует отметить, что 1H/14N обычно занимает менее 10 минут, потому что оба 1Рука 14N — многочисленные ядра.
Все вышеперечисленные измерения были выполнены с использованием спектрометра JNM-ECZ600R (JEOL RESONANCE Inc., Япония), оснащенного 1 мм быстрым зондом MAS ssNMR (JEOL RESONANCE Inc.) при 14.1 Тл. Количество образца, используемого в измерениях, составляет около 1 мг для каждого .
Fig.1
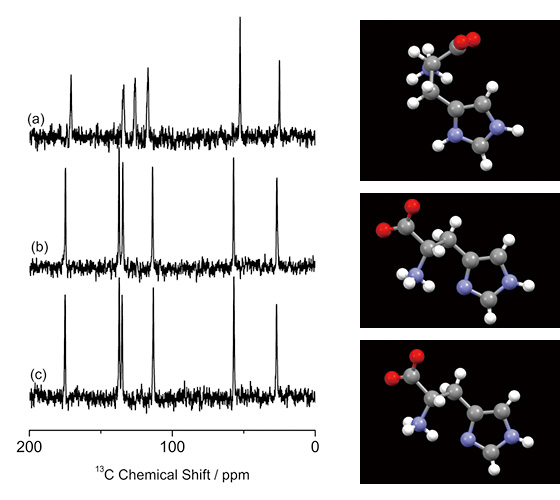
13C Спектры CPMAS (a) L-гистидин·HCl·H2O, (b) L-гистидин (орторомбический) и (c) L-гистидин (моноклинный) вместе с молекулярными конформациями. Было накоплено 256 сканов для (а) и 512 сканов для (б) и (в). (а) измерено при 16.4 Тл на спектрометре JNM-ECA700II (JEOL RESONANCE Inc., Япония). Рисунок воспроизведен со ссылки 7.
Fig.2

(а) ПЭМ-изображение и (б) картина дифракции нанопучка (диаметром 100 нм) микрокристаллического L-гистидина·HCl·H2О образец. Дифрактограмма была получена из области, обозначенной белым кружком на (а). Рисунок воспроизведен со ссылки 7.
Fig.3
( а ) Отрицательно отображаемая экспериментальная картина ED для L-гистидина (орторомбическая) и рассчитанная картина ED для ( b ) L-гистидина ( орторомбическая ) и ( c ) L-гистидина (моноклинная ). Расстояния d, соответствующие дифракционным пятнам, оцениваются по длине калиброванной камеры 40 см и длине волны 2.51 пм при ускоряющем напряжении 200 кВ. Картина ЭД рассчитывалась для пучка, падающего по оси зоны [631]. Рисунок воспроизведен со ссылки 7.
Fig.4

( а ) Отрицательно отображаемая экспериментальная картина ED для L-гистидина (моноклинная) и рассчитанная картина ED для ( b ) L-гистидина (орторомбическая) и ( c ) L-гистидина (моноклинная). Расстояния d, соответствующие дифракционным пятнам, оцениваются по длине калиброванной камеры 40 см и длине волны 2.51 пм при ускоряющем напряжении 200 кВ. Картина ЭД рассчитывалась для пучка, падающего по оси зоны [100]. Рисунок воспроизведен со ссылки 7.
Fig.5
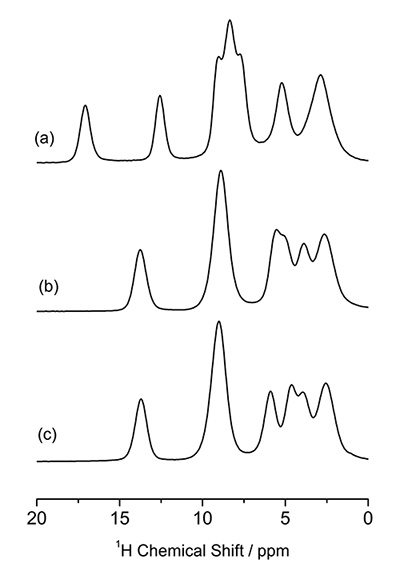
Ассоциация 1Спектры ЯМР H (а) L-гистидин·HCl·H2O, (b) L-гистидин (орторомбический) и (c) L-гистидин (моноклинный). Рисунок воспроизведен из ссылки 7.
Fig.6
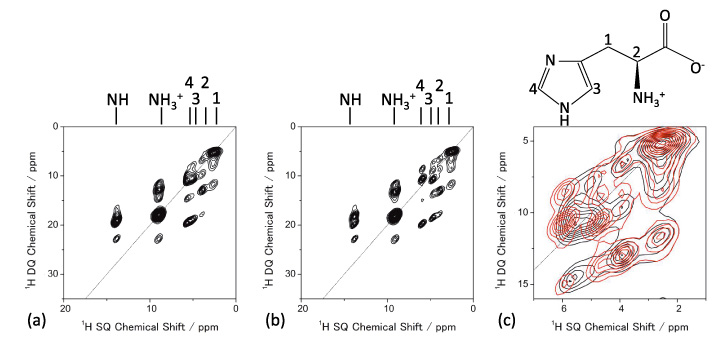
1Н ДК/1Спектры ЯМР H SQ (а) L-гистидина (орторомбический) и (б) L-гистидин (моноклинный). Расширение наложенных спектров также показано на (c) (L-гистидин (ромбический) черным цветом и L-гистидин (моноклинный) красным цветом). Четыре сканирования для каждого t1 периода накоплено 32 т1 приращения. Рисунок воспроизведен со ссылки 7.
Fig.7
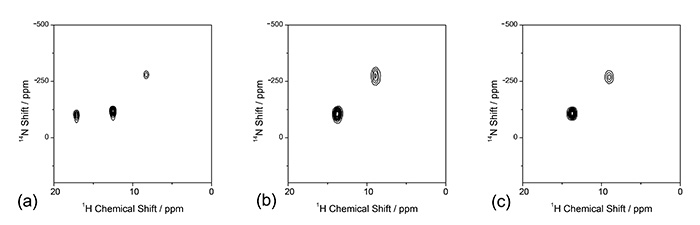
Ассоциация 1ЧАС/{14N} Спектры ЯМР (а) L-гистидин·HCl·H2O, (b) L-гистидин (орторомбический) и (c) L-гистидин (моноклинный). Спектр (а) был измерен при частоте MAS 90 кГц с помощью зонда HXMAS 0.75 мм (JEOL RESONANCE Inc., Япония). Восемь сканирований для каждого t1 периода были накоплены с (a) 64 и (b)/(c) 32 т1 приращения. Нет 1ЧАС-14Повторное связывание азота применяли во время перемешивания. Рисунок воспроизведен со ссылки 7.
Новые методы, разработанные для кристаллических полиморфов
Несмотря на важность азота в химии, фармации, материаловедении и т. д., ЯМР азота весьма ограничен. Просто потому, что низкая чувствительность 15N ЯМР из-за низкого содержания 15Н (0.4%). Несмотря на то что 15N предпочтительнее из-за его спинового квантового числа I = 1/2, другого изотопа 14N также является ЯМР-активным ядром с благоприятным высоким содержанием 99%. Целочисленное спиновое квантовое число (I = 1) и наличие квадрупольной связи затрудняет применение 14Н ЯМР. Для преодоления этих проблем мы разработали 1ЧАС-{14N} измерения гетероядерной многоквантовой когерентности (HMQC) в условиях очень быстрой MAS [8]. Этот метод обеспечивает высокопроизводительные измерения с образцами с ограниченной массой менее 1 мг. Из-за высокой чувствительности метод может быть расширен до трех измерений для 14N/1Н ДК/1Эксперименты по корреляции H SQ[9]. Кроме того, метод исследует наблюдение за 35Cl, который часто встречается в фармацевтической соли [10].
Отнесение сигналов помогает интерпретировать спектры ЯМР, как обсуждалось выше на рис. 6. В то время как двумерная гомо/гетероядерная корреляция очень помогает относить сигнал через двухспиновые коннективы, трехмерные эксперименты дают гораздо более четкие отнесения. С этой целью мы разработали 13C/1Н ДК/1Корреляция H SQ для выявления локальных 1H-1H-спиновая сеть вблизи 13С [11]. Полные назначения были получены из одного трехмерного спектра с образцом 1 мг при естественном содержании.
Соль/Сокристалл/Континуум[12]
Как обсуждалось выше, идентификация соли/сокристалла/континуума все еще остается большой проблемой в фармацевтических применениях из-за отсутствия возможности определить местонахождение водорода с помощью XRD. Различие между этими тремя классами можно интерпретировать как разные расстояния между водородом и азотом (рис. 8). Таким образом, размер 1H-15N дипольных взаимодействий, обратно пропорциональных кубу межъядерных расстояний, должны дать четкий ответ на вопрос соль/сокристалл/континуум.
Здесь мы предлагаем комбинированный метод SCXRD и ssNMR. В то время как SCXRD определяет глобальную кристаллическую структуру, ssNMR дает локальные межъядерные расстояния. Объединив эти два вида информации, можно установить цельное понимание кристаллической структуры. В качестве демонстрации сначала мы синтезируем четыре модельные многокомпонентные системы, показанные на рис. 9. Кристаллические структуры определяются с помощью SCXRD. Результаты ясно показывают наличие межмолекулярного контакта между N и OH, как и ожидалось (рис. 10). Вопрос в том, является ли это солью/сокристаллом/континуумом. Чтобы ответить на этот вопрос, мы попытались определить положения водорода с помощью SCXRD, однако он дает значения, зависящие от машины, что приводит к ненадежным положениям. Как ясно показывает кристаллическая структура, определенная с помощью SCXRD, существует межмолекулярное взаимодействие 1H/15N. 1H/15N расстояний, таким образом, сила связи между 1Рука 15N, должен дать ответ на проблемы с солью/сокристаллом/континуумом. Например, 1H/15Расстояние N в SA2 определено равным 1.25 Å (рис. 10). При расстояниях между азотом и кислородом 2.54 Å, определенных с помощью SCXRD, мы можем заключить, что SA2 является континуумом, поскольку водород расположен посередине кислорода и азота. Следует отметить, что SA2 состоит из сильной кислоты и основания с ΔpKa больше 3. Этот результат подчеркивает важность измерений SCXRD/ssNMR для идентификации соли/сокристалла/континуума даже для системы с большим ΔpKa. Структуры и межъядерные расстояния остальных трех систем были успешно определены (рис. 10). Узким местом этого метода является экспериментальное время для 1H-15N измерение расстояния. Помимо низкой естественной численности 15N и малый 1H-15N, эти фармацевтические образцы обычно показывают очень 1HT1 время отдыха. Кроме того, потребность в быстрой МАС уменьшает объем пробы, что приводит к дальнейшему снижению чувствительности. Мы разработали метод повышения пропускной способности, как описано в следующем разделе, каждое измерение по-прежнему требует 4-5 дней. Мы согласны с фактом ограниченной пропускной способности. Тем не менее, мы считаем, что этот метод весьма полезен, так как ни один другой метод не дает четкого ответа.
Все измерения ЯМР проводились с помощью ЯМР-спектрометра JNM-ECA700II (JEOL RESONANCE Inc.) с использованием 1-мм двойного резонансного быстрого датчика MAS (JEOL RESONANCE Inc.) при 16.4 Тл.
Fig.8
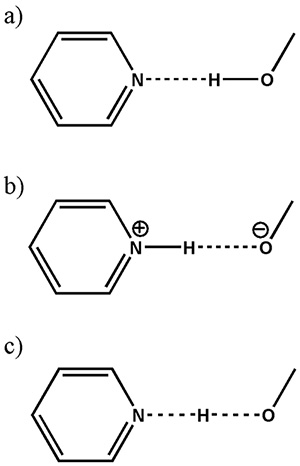
Схематическое изображение (а) сокристалла, (б) соли и (в) континуума (где положение атома Н является общим для двух тяжелых атомов) в типичном O...H...Н взаимодействие. Рисунок воспроизведен из ссылки 12.
Fig.9
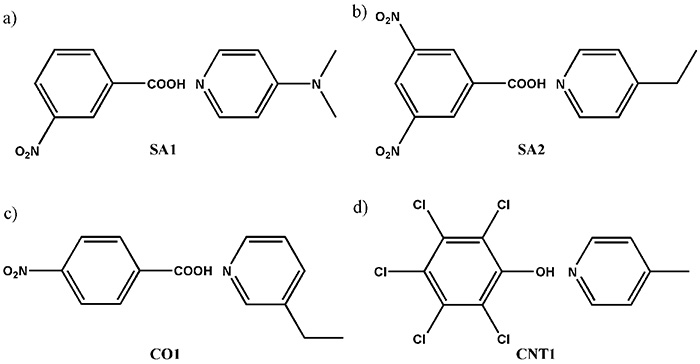
Схематическое изображение соединений, используемых в настоящем исследовании, показывающее (a) SA1 (3-нитробензойная кислота и N,N-диметилпиридин-4-амин), (b) SA2 (3,5-динитробензойная кислота и 4-этилпиридин), (c) CO1 (4-нитробензойная кислота и 3-этилпиридин) и (d) CNT1 (пентахлорфенол и 4-метилпиридин). Рисунок воспроизведен из ссылки 12.
Fig.10
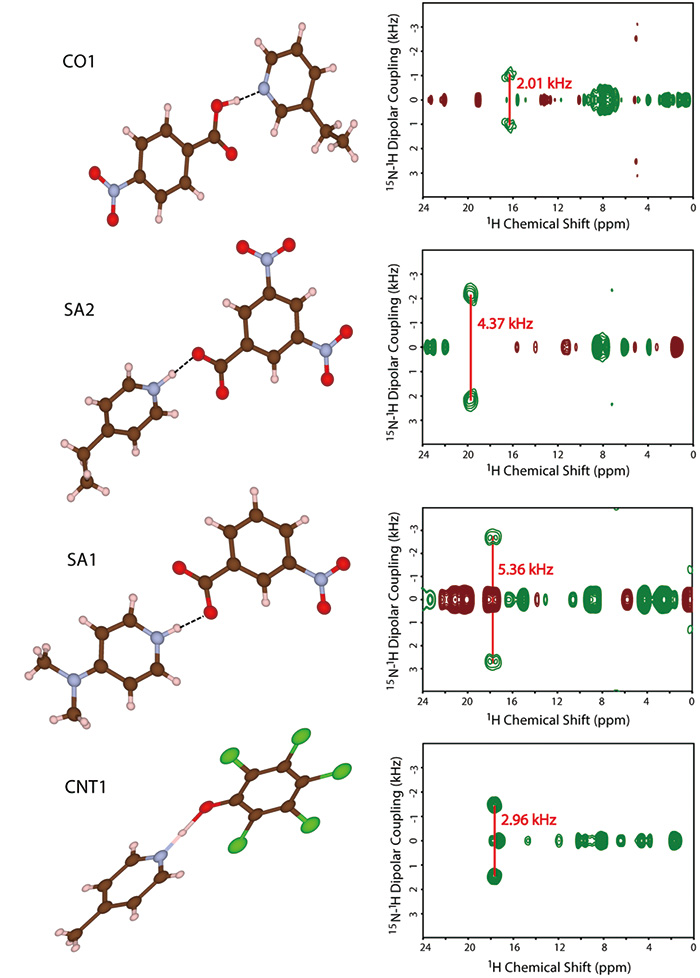
Молекулярные структуры и двумерные спектры invCP-VC (15N-1H дипольные связи по сравнению с 1H химический сдвиг) SA1, SA2, CO1 и CNT1. Рисунок воспроизведен из ссылки 12.
Разработаны новые методы для соли/сокристалла/континуума
Размеры 1H/15N дипольное взаимодействие должно быть точно измерено. Однако это непростая задача из-за 1) низкой численности 15N (0.4%), 2) малое гиромагнитное отношение (1/10 от 1H) и 3) обильный 1Ядра H, которые не связаны с 15N, что приводит к низкой чувствительности, небольшой дипольной связи (1-7 кГц) и перекрытию 1H резонансов соответственно. Что еще более важно, предыдущие методы, как правило, показывают 1H/15N дипольная связь, которая зависит от условий эксперимента, дает ненадежные расстояния. Чтобы преодолеть эти трудности, мы внедрили метод обратного детектируемого КП с переменным временем контакта (инв КП-ВК) при быстром МАС [13]. Во-первых, метод дает хорошо выраженные дипольные расщепления, не зависящие от условий эксперимента [14]. На самом деле масштабный коэффициент метода inv CP-VC в значительной степени зависит от напряженности радиочастотного поля. Однако сигналы из области с B1 неоднородности рассеиваются в широком спектральном диапазоне, что делает эти сигналы невидимыми. Таким образом, сигналы от однородных B1 поля наблюдаются избирательно. Повышение чувствительности за счет 1Обнаружение H в inv CP-VC смягчает низкую чувствительность из-за небольшого содержания и небольшого объема пробы. Экспериментально показано, что inv CP-VC способен контролировать 1H/15N связь 2 кГц. Inv CP-VC работает как фильтрация 1H-резонансы, обеспечивающие выборочное наблюдение 1Н в непосредственной близости от 15N. Это позволяет избежать сложности перекрывающихся резонансов между 1Хс. Кроме того, inv CP-VC позволяет анализировать образцы с ограниченной массой менее 1 мг.
Задержка повторения должна быть вставлена между последовательными сканированиями в измерениях ЯМР, чтобы спиновая система возвращалась близко к тепловому равновесию перед началом измерений ЯМР. Поскольку задержка повторения должна быть порядка T1 время релаксации, которое обычно намного больше, чем в остальных экспериментах, T1 время релаксации является доминирующим фактором времени эксперимента в ЯМР. В экспериментах inv CP-VC намагниченность начинается с 1Н, таким образом, 1HT1 является важным. Однако хорошо кристаллические низкомолекулярные АФС, как правило, имеют очень длительный срок службы. 1HT1 время релаксации от десятков до сотен секунд, что резко снижает пропускную способность. При высокой скорости MAS проблема более серьезная, потому что 1H-1Диффузия спинов H подавляется, что приводит к более длительному 1HT1 время отдыха. Поскольку интересующие нас протоны NH пространственно и спектрально изолированы от других 1H, очень медленные 1H-1Спиновая диффузия H препятствует 1Намагниченность H от восстановления до своего теплового равновесия. Это приводит к более медленному 1HT1 время релаксации протонов NH по сравнению с остальными 1Хс. Чтобы преодолеть эту трудность, мы применили последовательности повторной связи, управляемой радиочастотой (RFDR), на 1H, во время задержки повторения [15]. Это повышает 1H-1Спиновая диффузия H, выводящая намагниченность из быстро релаксирующей 1H в NH протоны. Поскольку механизмы релаксации быстро релаксирующих протонов используются несколько раз, можно улучшить не только протон NH, но и общую чувствительность. Мы также оптимизировали фазовое циклирование в последовательности RFDR, подходящее для 1H смешение [16, 17].
Заключение
И ПЭМ, и ЯМР постоянно совершенствуются даже сейчас, несмотря на то, что они были введены более 70 лет назад. Недавно представленная высокочувствительная камера в ПЭМ открывает путь к новому применению в крио-ПЭМ для определения структуры белка с использованием подхода к анализу отдельных частиц. Повышение чувствительности полезно не только для белков, но и для образцов, чувствительных к лучу, включая низкомолекулярные АФИ. Еще один большой всплеск наблюдается в быстрой технологии MAS ssNMR, которая позволяет 1H ЯМР даже жестких твердых веществ. Эти новые разработки не только предоставляют новую информацию, которая ранее была недоступна, но и повышают пропускную способность, что является одним из ключевых факторов широкого распространения использования. Кроме того, очень важно применять несколько различных аналитических методов, дополняющих друг друга. Правильная комбинация обеспечивает полезную информацию с высокой пропускной способностью. Действительно, JEOL Ltd. также продвигает стратегию YOKOGUSHI, предлагая привлекательные комбинации оборудования.
В этой статье мы объединяем XRD, ED и ssNMR для решения структурной проблемы в фармацевтических науках, включая кристаллические полиморфы и проблемы соли/сокристалла/континуума. Для проблемы кристаллических полиморфов мы демонстрируем комбинированный подход ЭД и 1H быстрый MAS ЯМР для идентификации кристаллической формы в качестве метода, дополняющего PXRD и 13С КПМАС. ЭД позволяет определять кристаллическую форму монокристаллов от нано- до микроразмеров. С другой стороны, 1H быстрый MAS может различать кристаллическую форму с аналогичной молекулярной конформацией. Кроме того, быстрый MAS позволяет 1H/14Корреляции N, дающие положения протонов, близкие к ядрам азота. Для идентификации соли/сокристалла/континуума вводится комбинированный подход SCXRD и ssNMR при очень высокой скорости MAS. Хотя SCXRD обеспечивает глобальную молекулярную структуру, он не может определить положения протонов, которые имеют решающее значение для проблемы соли/сокристалла/континуума. С другой стороны, ссЯМР дает точные 1H-15N расстояний, хотя глобальная структура получается с трудом. Этот подход обеспечивает четкое понимание молекулярной структуры от кристаллической структуры до водородных связей, чтобы ответить на проблему соли/сокристалла/континуума. Формирование континуума в многокомпонентных системах с ΔpKa > 3 подчеркивает важность тщательного исследования с помощью SCXRD и ssNMR.
Благодарности
Все представленные здесь работы были выполнены совместно с членами нашей группы в коллаборационной лаборатории RIKEN CLST-JEOL. Я хотел бы поблагодарить всех участников группы и нашего коллегу из JEOL Ltd. и JEOL RESONANCE Inc. за огромную поддержку. Я также хотел бы поблагодарить наших друзей из Индийского института наук в Бангалоре, лаборатории AMES, Киотского университета, Токийского сельскохозяйственного и технологического университета, Уорикского университета, Лилльского университета, Мичиганского университета за плодотворное сотрудничество.
Рекомендации
- РК Харрис, Дж. Фарм. Фармакол. 59 (2007) 225 239–XNUMX.
- М. Геппи, Г. Моллика, С. Борзакки, К. А. Верачини, заявл. Спектроск. Преподобный. 43 (2008) 202 302–XNUMX.
- ФГ Фогт, Будущее мед. Химия. 2 (2010) 915 921–XNUMX.
- Ю. Нишияма*, Твердотельный ЯМР с вращением образцов под магическим углом, частота 60–100 кГц для образцов естественного изобилия, Ядерно-магнитный резонанс твердого тела, 78 (2016) 24-36. DOI: 10. 1016/j. сснмр. 2016. 06. 002.
- Ю. Нишияма*, ЯМР твердого тела при сверхвысокой частоте MAS 40–120 кГц, в книге «Экспериментальные подходы к ЯМР-спектроскопии – методология и применение в науках о жизни и материаловедении», Springer (2017).
- Т. Кобаяши, Ю. Нишияма*, М. Пруски*, Гетероядерная корреляционная спектроскопия с обратным детектированием, Современные методы ЯМР твердого тела: Руководство для практиков, Королевское химическое общество (2018).
- Т. Ойкава, М. Окумура, Т. Кимура, Ю. Нишияма*, ЯМР твердого тела в сочетании с электронной дифракцией: определение кристаллических полиморфов небольших органических микрокристаллических образцов, Акта Крист. C73 (2017) 219–228. DOI: 10. 1107/S2053229617003084.
- Ю. Нишияма*, Ю. Эндо, Т. Немото, Х. Уцуми, К. Ямаути,
Вы медицинский работник или персонал, занимающийся медицинским обслуживанием?
Нет
Напоминаем, что эти страницы не предназначены для предоставления широкой публике информации о продуктах.